17 October
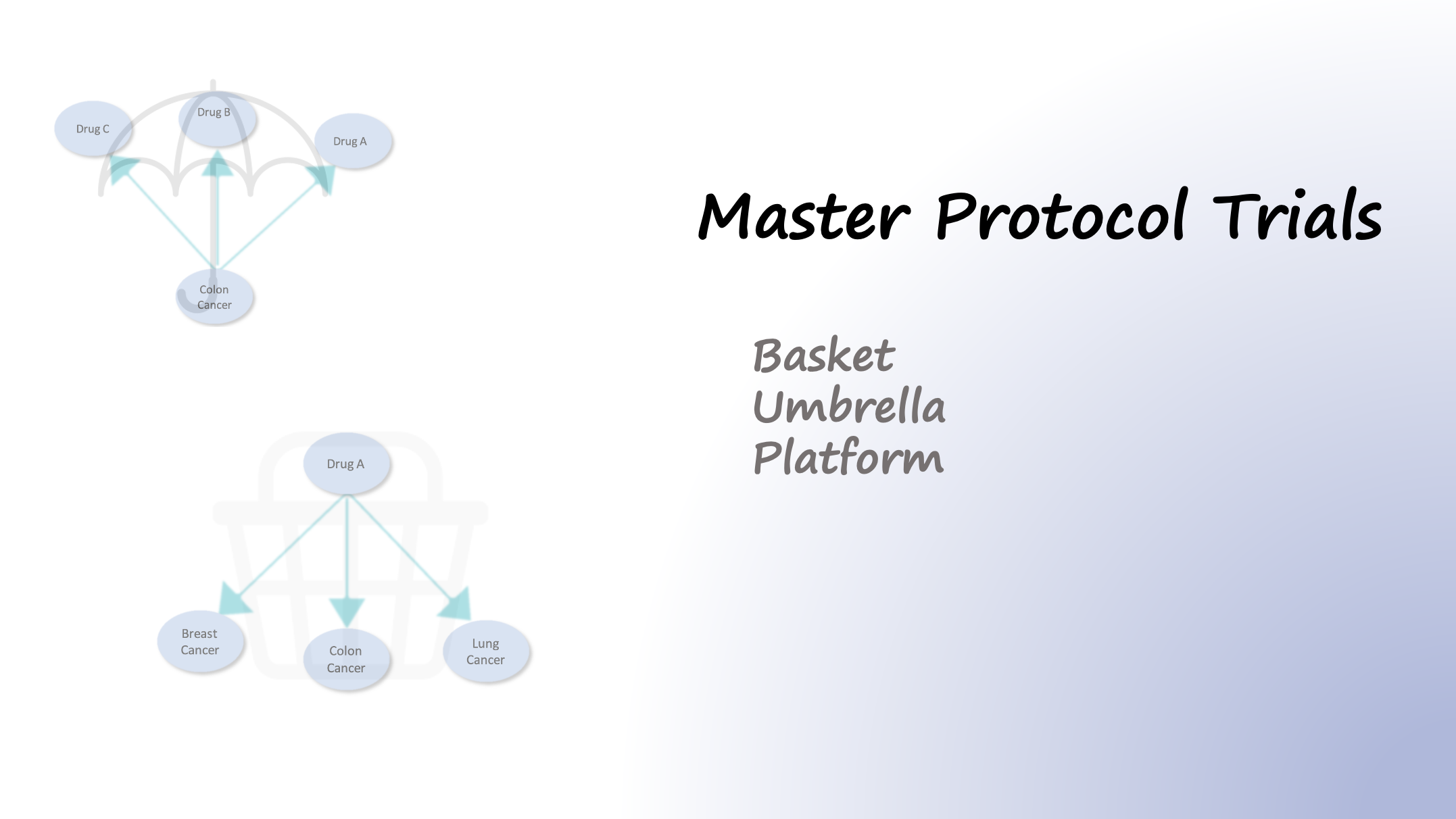
Just by looking at the following infographic published by cbinsights, you can see a patient’s journey who is looking for clinical studies, after all forms of available treatments have failed to work. Now let’s take a look at the structure of a clinical site and see how the AI can facilitate…