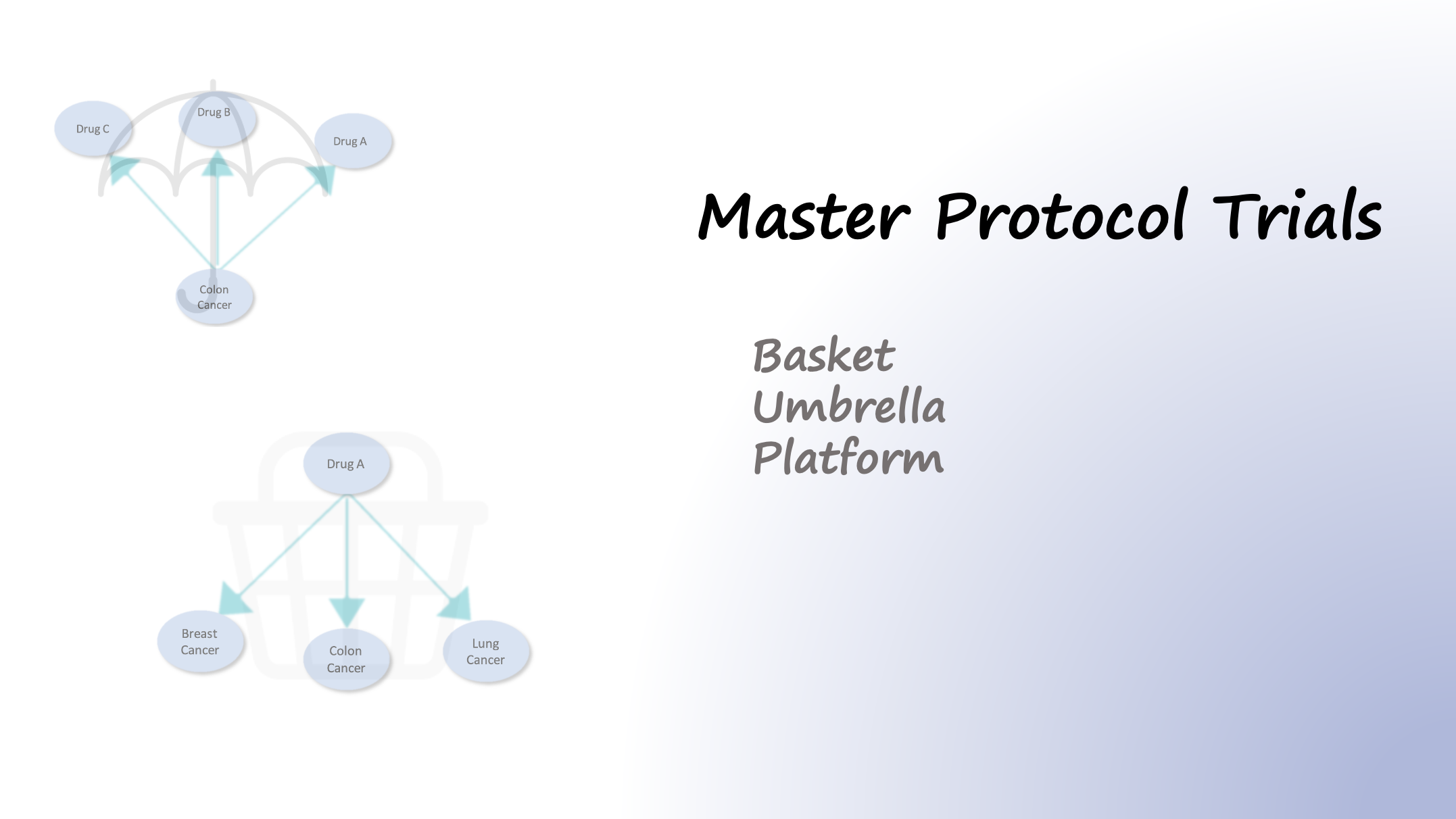
05 January
Feasibility assessment is a critical step in conducting a successful clinical trial. Selecting the right site can be beneficial for both the sponsor and the clinical site. In addition, it can provide an opportunity for many patients to be enrolled in a study that may change their life. Therefore, the…