
28 October
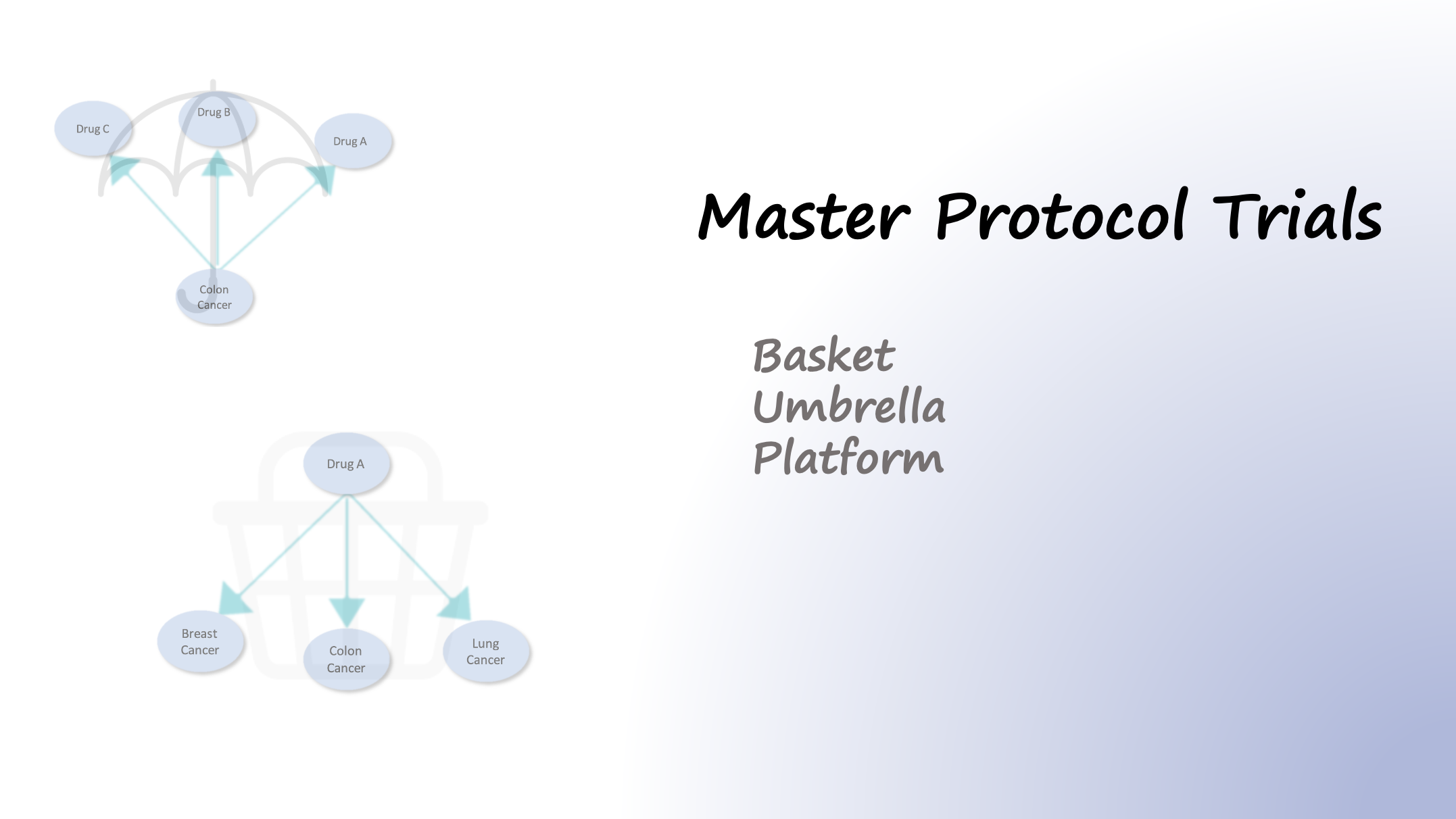
In traditional trial designs, a single drug is tested in a single disease population in one clinical trial. In contrast to traditional trial designs, master protocols, use a single infrastructure, trial design, and protocol to simultaneously evaluate multiple drugs and/or disease populations in multiple substudies, allowing for efficient and accelerated…




